
GM1 Gangliosidosis: A History
From First Clinical Descriptions to Modern Therapeutics
Published July 2026
Part 1: Clinical History of GM1
What we now call GM1 gangliosidosis spent nearly a century hiding in plain sight, misclassified under a diagnostic umbrella so broad it concealed multiple distinct diseases. Its story is inseparable from the story of Tay-Sachs disease, and from the slow development of the biochemical tools needed to tell them apart. The following is a rich history of GM1 gangliosidosis.
The First Observations: Tay and Sachs (1881-1890s)
In 1881, the British ophthalmologist Warren Tay published a case report describing an infant with progressive neurological deterioration and a distinctive cherry-red spot at the macula.[1] The following decade, the American neurologist Bernard Sachs described a series of similar cases, which he termed “amaurotic familial idiocy.”[2]
Sachs recognized the familial pattern and the characteristic pathological findings in neurons, and his terminology became the standard diagnostic label for an entire class of childhood neurodegenerative diseases characterized by progressive neurological regression, vision loss, and early death.
The phrase “amaurotic familial idiocy” was expansive by design. It described a phenotype, not a biochemical entity. Any child with neurological regression, visual involvement, and a suggestive family history could be placed under this label. This diagnostic flexibility would prove consequential for decades.
The Discovery of Gangliosides (1935-1942)
In the 1930s and early 1940s, the German biochemist Ernst Klenk, working at the University of Cologne, identified a class of complex lipids enriched in the brain and found at markedly elevated levels in the stored material of patients with the diseases then called “amaurotic familial idiocy.” He named these compounds gangliosides, from the Greek for ganglion, reflecting their abundance in neural tissue.[3] Klenk’s work established that these were lipid storage diseases at the biochemical level, but the tools to distinguish between different ganglioside subtypes did not yet exist.
Clinical Separation Begins (1959-1964)
When we look at the history of GM1 gangliosidosis, the first clinical hints that the diseases collected under the amaurotic idiocy umbrella were not all the same began to emerge in the late 1950s and early 1960s. R.M. Norman and colleagues published observations on a subset of patients with neurological deterioration who also exhibited visceral involvement, including hepatosplenomegaly and skeletal dysplasia, features not typical of classical Tay-Sachs disease.[4]
In 1964, Benjamin Landing and colleagues described what they called “familial neurovisceral lipidosis,” characterizing the systemic nature of the storage in eight patients previously misclassified as having Hurler syndrome variants or Tay-Sachs disease with visceral involvement.[5] This was among the first clinical descriptions that would eventually be recognized as GM1 gangliosidosis. The broader category “generalized gangliosidosis” was used to distinguish these cases from the neurologically confined presentation of classical Tay-Sachs.
Ganglioside Nomenclature (1963)
A critical intellectual tool arrived with the systematic ganglioside nomenclature developed by the Swedish biochemist Lars Svennerholm in 1963. Svennerholm’s system used a letter-number code (GM1, GM2, GM3, GD1a, etc.) to denote the sugar chain structure and sialic acid content of individual ganglioside species.[6] For the first time, researchers could specify which ganglioside was accumulating in which disease. This nomenclature revealed that the storage material in classical Tay-Sachs was predominantly GM2 ganglioside, while the material in the “generalized” cases was predominantly GM1. The diseases had different substrates. The question became: why?
The Enzymatic Solution (1968-1969)
The definitive biochemical separation came in two landmark publications from the same laboratory at the University of California San Diego. In 1968, Yoshiyuki Okada and John S. O’Brien demonstrated that the enzymatic defect in generalized gangliosidosis was a profound deficiency of beta-galactosidase, the lysosomal enzyme responsible for cleaving the terminal galactose from GM1 ganglioside.[7]
The following year, in 1969, the same team showed that the defect in classical Tay-Sachs disease was a different enzyme entirely: hexosaminidase A.[8] Two diseases, two enzymes, two genes: the distinction was now absolute. Approximately 87 years had elapsed since Warren Tay’s first case report.
Molecular Genetics and Subtype Classification (1970s-1990s)
Following the enzymatic discoveries, researchers characterized the clinical spectrum more precisely. Three subtypes of GM1 gangliosidosis were recognized based on age of onset and rate of progression:
- Type 1 (infantile): Onset in the first six months of life, severe neurological regression, hepatosplenomegaly, skeletal changes, cherry-red spot in approximately 50% of cases. Most affected children do not survive beyond age 2-3 years.
- Type 2 (late-infantile/juvenile): Onset between 7 months and 3 years, progressive motor and cognitive deterioration, less severe visceral involvement. Survival into the first or second decade is possible, though the disease is ultimately fatal.
- Type 3 (adult/chronic): Onset from adolescence onward, predominantly movement disorder (dystonia, ataxia, parkinsonism) with relative sparing of cognition in some patients. The course is variable and can extend decades.
In terms of the timeline for the history of GM1 gangliosidosis, the molecular basis was fully established in the early 1990s with the cloning of the GLB1 gene and systematic identification of pathogenic variants. GLB1 mutations are inherited in an autosomal recessive pattern. Hundreds of distinct pathogenic variants have now been catalogued, and genotype-phenotype correlations, while imperfect, provide some predictive value for clinical course.
Part II: Why GM1 Was Mistaken for Tay-Sachs for Nearly a Century
Given that GM1 gangliosidosis and Tay-Sachs disease are caused by defects in different genes, different enzymes, and different substrates, the near-century of conflation requires explanation. Six structural factors account for it.
1. Phenotypic Overlap at the Clinical Level
Both diseases present in infancy with progressive neurological deterioration, loss of developmental milestones, hypotonia, and seizures. The cherry-red macular spot, while more consistently present in Tay-Sachs, also appears in a substantial proportion of GM1 Type 1 patients. Before enzymatic and genetic testing existed, a clinician confronting an infant with these features had no reliable way to distinguish between the two conditions. The clinical presentations were, from the bedside, largely indistinguishable in the severe infantile forms.
2. The “Amaurotic Familial Idiocy” Diagnostic Umbrella
Sachs’s terminology described a phenotypic category rather than a single disease entity.[2] The label was applied broadly to any child with the relevant constellation of findings, without the expectation that it represented a single biochemical disorder. Because the category was defined clinically and was explicitly intended to capture a family of related presentations, there was no conceptual pressure to subdivide it until biochemical tools made subdivision possible.
3. The Absence of Lysosomal Biology as a Framework
The concept of the lysosome as a discrete cellular organelle with a specific complement of hydrolytic enzymes was not established until Christian de Duve’s work in the 1950s, for which he received the Nobel Prize in Physiology or Medicine in 1974.[9] The idea that a disease could result from the loss of a specific lysosomal enzyme was not conceptually available to clinicians and pathologists working in the first half of the twentieth century. Without this framework, the biochemical individuality of different ganglioside storage diseases could not be recognized or sought.
4. Ganglioside Chemistry Was Not Yet Resolved
Even after Klenk identified gangliosides as the stored material in these diseases,[3] the analytical chemistry needed to distinguish between individual ganglioside species was not available until the 1960s. Thin-layer chromatography and the systematic nomenclature developed by Svennerholm[6] were prerequisites for recognizing that different patients were accumulating different gangliosides. Until those tools existed, the biochemical distinction between GM1 and GM2 storage was not demonstrable, even in principle.
5. Enzyme Assay Technology Arrived Late
The direct measurement of lysosomal enzyme activity in patient samples was not technically practical until the mid-to-late 1960s. The assays developed by Okada, O’Brien, and their contemporaries[7][8] required synthetic fluorogenic substrates and spectrofluorometric equipment that did not exist in earlier decades. The enzymatic diagnoses of Tay-Sachs and GM1 gangliosidosis were simultaneous developments, not because researchers had been uninterested in the question but because the technical means to answer it had only just become available.
6. Shared Inheritance Pattern
Both conditions are autosomal recessive, are more prevalent in certain founder populations, and tend to present with familial clustering. The similar epidemiological and inheritance patterns provided no discriminating signal at the clinical level, reinforcing the perception of a single disease category.
In summary, GM1 gangliosidosis and Tay-Sachs disease are phenotypically similar in their severe forms, were grouped under an intentionally broad diagnostic label, and could not be biochemically distinguished until the convergence of lysosomal biology, ganglioside chemistry, and enzyme assay technology in the 1960s. The approximately 87-year delay was not a failure of clinical observation but a reflection of the state of biochemical science.
Part III: The Modern Era (1990s to Present)
Following molecular characterization of the GLB1 gene and the cataloguing of pathogenic variants, the field turned to understanding the natural history of the disease in sufficient detail to support therapeutic development, and to identifying and testing candidate treatments.
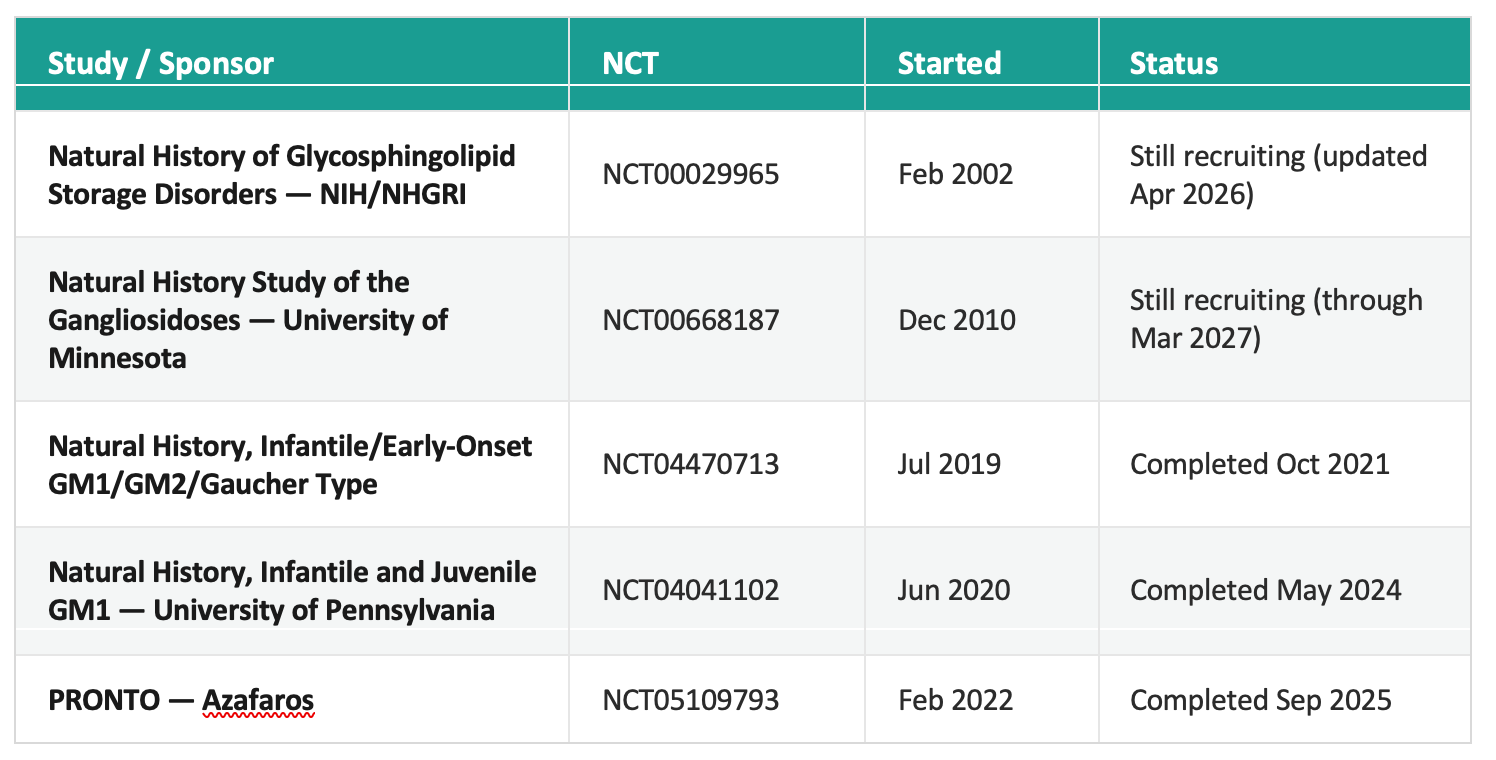
Natural History Studies
Systematic natural history studies were essential prerequisites for clinical trial design, providing the outcome measures, progression rates, and patient stratification frameworks needed to evaluate any intervention. The major studies are summarized below.
Substrate Reduction Therapy
Substrate reduction therapy (SRT) aims to reduce the rate of glycosphingolipid synthesis, decreasing the substrate load that the deficient lysosomal enzyme cannot clear. Three agents have been investigated in the context of GM1 gangliosidosis.
Miglustat (Zavesca)
Miglustat, an oral inhibitor of glucosylceramide synthase, was the first SRT agent used off-label in patients with GM1 and GM2 gangliosidoses. A formal clinical trial at the University of Minnesota (NCT02030015, initiated December 2015) evaluated miglustat in GM1/GM2 patients but was terminated before completion. Off-label use outside of formal trials has been reported by several treating centers.
Venglustat (Ibiglustat) and the AMETHIST Trial
Venglustat is a more potent and CNS-penetrant glucosylceramide synthase inhibitor developed by Sanofi. The pivotal AMETHIST trial (NCT04221451) was a Phase 3 study initiated June 29, 2020. The trial had two components. The primary arm enrolled adults with late-onset GM2 gangliosidosis (Tay-Sachs and Sandhoff disease) in a randomized, placebo-controlled design.
A secondary open-label basket arm enrolled 16 patients aged 2 years and older with related conditions, including 7 patients with GM1 gangliosidosis. Results for the GM1 basket arm, published by Tifft and colleagues in Genetics in Medicine,[13] showed meaningful biomarker reductions: CSF glucosylceramide decreased by 70.2% and CSF GM1 ganglioside decreased by 29.1%.
FARS-neuro scores improved and participants remained clinically stable during the observation period. One late-infantile GM1 participant enrolled at age 2 died from disease progression during the study. In the primary arm, CSF GM2 was significantly reduced in adults with late-onset GM2, but no improvement was observed on functional endpoints. The AMETHIST trial was terminated following this result.
Nizubaglustat (AZ-3102) and the Azafaros Phase 3 Program
Azafaros is developing nizubaglustat, a next-generation GCS inhibitor, in dedicated Phase 3 trials for late-infantile and juvenile GM1 and GM2 gangliosidosis. Two trials launched in June 2025: NCT07054515 (GM1) and NCT07082543 (GM2). These represent the first Phase 3 trials designed with GM1 gangliosidosis as a primary indication.
AAV Gene Therapy
Gene therapy for GM1 gangliosidosis introduces a functional copy of GLB1 via an adeno-associated virus (AAV) vector. Preclinical foundations were established by Miguel Sena-Esteves and colleagues at the University of Massachusetts Chan Medical School, who developed an AAV9-GLB1 vector and demonstrated efficacy in murine and naturally-occurring feline models of the disease.[10][11] Several programs have entered the clinic.
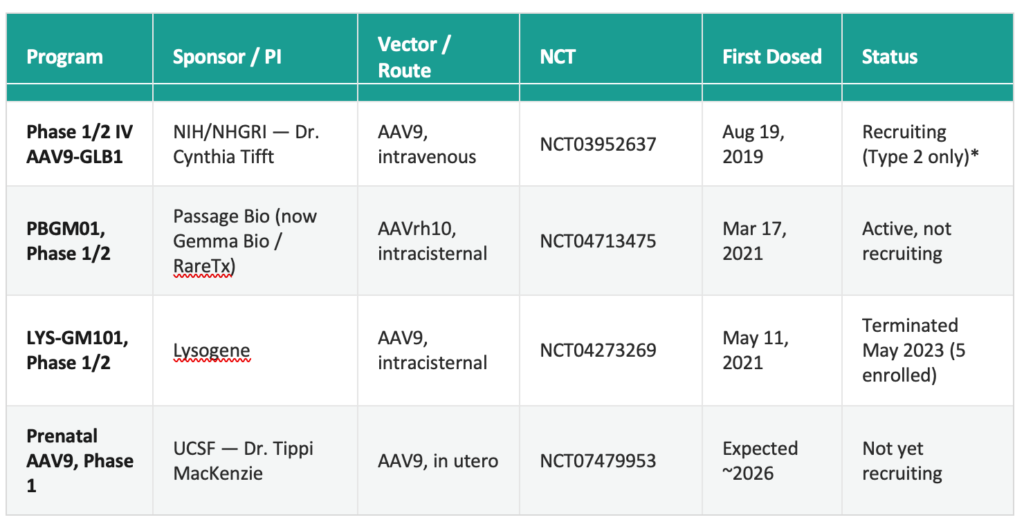
* ClinicalTrials.gov continues to list both Type 1 and Type 2 eligibility for NCT03952637, but recruitment has in practice been restricted to Type 2 patients. The registry entry has not been updated to reflect this. Results from the first nine enrolled children were published in the New England Journal of Medicine.[12]
The NIH trial and the RareTx/UPenn next-generation AAVhu68 intracisternal program represent the most active investigational gene therapy efforts in GM1 gangliosidosis as of mid-2026. The liquidation of Lysogene in May 2023 and the dissolution of Sio Gene Therapies (formerly Axovant) in February 2024 reduced the number of active commercial programs, placing greater weight on academic and foundation-supported efforts.
Frequently Asked Questions
What is GM1 gangliosidosis?
GM1 gangliosidosis is a rare, inherited lysosomal storage disease caused by mutations in the GLB1 gene, which lead to a deficiency of the enzyme beta-galactosidase. Without enough of this enzyme, GM1 ganglioside builds up in cells throughout the body, especially in the brain, causing progressive neurological decline. The disease is inherited in an autosomal recessive pattern and occurs in three subtypes based on age of onset and severity.
What’s the difference between GM1 gangliosidosis and Tay-Sachs disease?
GM1 gangliosidosis and Tay-Sachs disease are distinct genetic disorders that were misclassified as the same condition for nearly 87 years because their severe infantile forms are almost indistinguishable at the bedside. GM1 is caused by a beta-galactosidase deficiency that leads to GM1 ganglioside accumulation, while Tay-Sachs is caused by a hexosaminidase A deficiency that leads to GM2 ganglioside accumulation. Researchers Yoshiyuki Okada and John O’Brien separated the two diseases enzymatically in 1968 and 1969.
What are the different types of GM1 gangliosidosis?
GM1 gangliosidosis has three recognized subtypes. Infantile (Type 1 ) begins in the first six months of life with severe neurological regression; most children do not survive past age 2-3. Late-infantile/juvenile (Type 2) begins between 7 months and 3 years, progresses more slowly, and allows survival into the first or second decade. Adult/Chronic (Type 3) begins in adolescence or later, primarily causing movement disorders such as dystonia and ataxia, with a variable course that can span decades.
Why did it take nearly a century to identify GM1 gangliosidosis as a separate disease from Tay-Sachs?
The two diseases weren’t distinguished until 1968-1969, about 87 years after Warren Tay’s first case report in 1881, because their clinical presentations overlap heavily and the biochemical tools needed to tell them apart didn’t exist yet. Only after the discovery of the lysosome in the 1950s, the development of ganglioside nomenclature by Lars Svennerholm in 1963, and the arrival of enzyme assay technology in the late 1960s could researchers show that GM1 and Tay-Sachs involve different enzymes, different substrates, and different genes entirely.
References
1. Tay W. Symmetrical changes in the region of the yellow spot in each eye of an infant. Trans Ophthalmol Soc UK. 1881;1:55-57.
2. Sachs B. On arrested cerebral development, with special reference to its cortical pathology. J Nerv Ment Dis. 1887;14:541-553. (NLM Historical Collection)
3. Klenk E. Über die Ganglioside, eine neue Gruppe von zuckerhaltigen Gehirnlipoiden [On gangliosides, a new group of sugar-containing brain lipids]. Hoppe-Seyler’s Z Physiol Chem. 1942;273:76-86.
4. Norman RM, Urich H, Tingey AH, Goodbody RA. Tay-Sachs’ disease with visceral involvement and its relationship to Niemann-Pick’s disease. J Pathol Bacteriol. 1959;78:409-421. PMID: 14427628.
5. Landing BH, Silverman FN, Craig JM, Jacoby MD, Lahey ME, Chadwick DL. Familial neurovisceral lipidosis: an analysis of eight cases of a syndrome previously reported as “Hurler-variant,” “pseudo-Hurler,” and “Tay-Sachs disease with visceral involvement.” Am J Dis Child. 1964;108:503-522. PMID: 14209687.
6. Svennerholm L. Chromatographic separation of human brain gangliosides. J Neurochem. 1963;10:613-623. PMID: 14066623.
7. Okada S, O’Brien JS. Generalized gangliosidosis: beta-galactosidase deficiency. Science. 1968;160(3831):1002-1004. PMID: 5647842.
8. Okada S, O’Brien JS. Tay-Sachs disease: generalized absence of a beta-D-N-acetylhexosaminidase component. Science. 1969;165(3894):698-700. doi:10.1126/science.165.3894.698.
9. de Duve C, Pressman BC, Gianetto R, Wattiaux R, Appelmans F. Tissue fractionation studies 6: intracellular distribution patterns of enzymes in rat-liver tissue. Biochem J. 1955;60(4):604-617. PMID: 13249955.
10. Weismann CM, Ferreira J, Keeler AM, et al. (Sena-Esteves M, senior author). Systemic AAV9 gene transfer in adult GM1 gangliosidosis mice reduces lysosomal storage in CNS and extends lifespan. Hum Mol Genet. 2015;24(15):4353-4364. PMID: 25964428.
11. Gross AL, Gray-Edwards HL, Bebout CN, et al. (Sena-Esteves M, co-senior author). Intravenous delivery of adeno-associated viral gene therapy in feline GM1 gangliosidosis. Brain. 2022;145(2):655-669. doi:10.1093/brain/awab309. PMID: 34410345.
12. Lewis CJ, D’Souza P, Johnston JM, Tifft CJ, et al. AAV9 gene therapy in type II GM1 gangliosidosis: a phase 1-2 trial. N Engl J Med. 2026 Mar 26;394(12):1184-1194. doi:10.1056/NEJMoa2510935. PMID: 41665410.
13. Tifft CJ, et al. Venglustat in GM2 gangliosidoses and related disorders: results of the AMETHIST randomized controlled and basket trials. Genet Med. 2025. doi:10.1016/j.gim.2025.101615. PMID: 41108138.
Prepared in collaboration with Cure GM1 Foundation. Trial status verified against ClinicalTrials.gov as of June 2026. Citations re-verified against PubMed and ClinicalTrials.gov July 2026.
This document does not constitute medical or legal advice.
Support Our Mission to Cure GM1
- Help fund critical research for a GM1 cure.
- Support families affected by GM1.
- Accelerate clinical trials for life-saving treatments.