
About GM1 Gangliosidosis
Everything you need to know:
What causes GM1 gangliosidosis, who it affects, how it’s inherited, and where research stands today.
What is GM1 Gangliosidosis?
GM1 gangliosidosis (GM1) is a rare, inherited lysosomal storage disorder caused by a mutation in the GLB1 gene. Without a functioning copy of this gene, cells cannot produce beta-galactosidase, an enzyme needed to break down GM1 ganglioside. This toxic buildup accumulates in neurons and other cells, causing progressive damage to the brain and nervous system. GM1 is fatal and currently has no approved cure or treatment. Children and adults with the disease face relentless neurological decline.

This video explains GM1 gangliosidosis:
The Cause

GM1 gangliosidosis is a lysosomal storage disorder (LSD). Lysosomes are the cell’s recycling system, responsible for breaking down and clearing waste molecules. When a lysosomal enzyme is missing or deficient, waste accumulates inside cells instead of being cleared, damaging cells and causing organ dysfunction. LSDs are individually rare but collectively affect a significant number of people worldwide.
The genetic lysosomal defect for those with GM1 gangliosidosis results in a deficiency of the enzyme beta-galactosidase. This enzyme is needed to break down and recycle a number of biomolecules. The substance that accumulates most in GM1 gangliosidosis is called GM1 ganglioside. Abundant in neurons, GM1 ganglioside plays important roles in the nervous system, including in cell signaling, neuronal protection, neuronal recovery, and apoptosis. Without beta-galactosidase to break it down, GM1 ganglioside accumulates to toxic levels in cells and eventually causes cell death, predominantly in neurons. When enough neurons die, the nervous system progressively deteriorates, which ultimately leads to death from complications of GM1 gangliosidosis.
References:
- NCBI Bookshelf: GM1 Gangliosidosis Overview
- FASEB Journal: GM1 Ganglioside and Lysosomal Storage
- Molecular Neurobiology: GM1 in Neurological Disease
Who Gets GM1 Gangliosidosis?
The inability to create a normally functioning form of the enzyme beta-galactosidase is due to a mutation in a gene called the GLB1 gene. This mutation is passed on via autosomal recessive inheritance. To understand autosomal recessive inheritance, it helps to understand the words that make up the term.
Autosomal
In genetics if a gene is autosomal, it means the gene is inherited in the same way by both males and females. In other words, the gene codes for a substance or characteristic that is not unique to male or female genders. The GLB1 gene is autosomal.
Recessive
Everyone has two copies of each gene, one from their mother and one from their father. Some genes are “dominant,” meaning the trait they code for is expressed regardless of what the other gene in the pair codes for. Some genes are “recessive,” meaning the trait is only expressed if both copies of the gene code for it in the same way.
Inheritance
This refers to something handed down from parents to children, and in genetics, that’s genes and traits. This means a person can only have GM1 gangliosidosis if both of their parents have a copy of the mutated gene and the copy of the mutated gene from each parent was passed down to their child.
Incidence of GM1
In the global population, only 1 in 250 people have a copy of the GLB1 gene with the disease-causing mutation. Their other GLB1 gene is normal. When a person only has one copy of the mutated GLB1 gene, they are called carriers. Carriers don’t have the disease but can pass down the mutated gene that causes it. Each carrier has a 50% chance of passing on the mutated gene. This means when both parents are carriers, their children have a 25% chance of inheriting two mutated GLB1 genes.
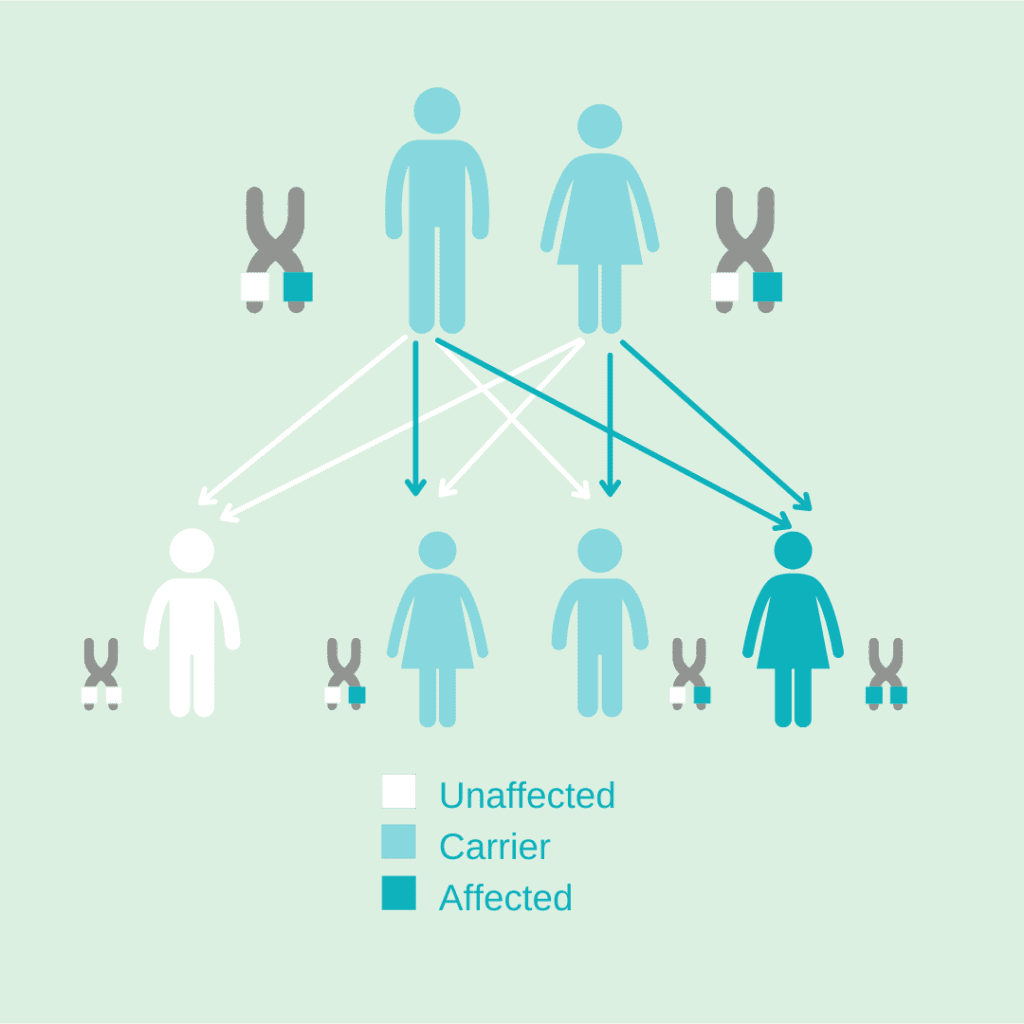
This is part of what makes GM1 gangliosidosis so rare. It is unlikely that two carriers have children, and if they do, they only have a 25% chance of having a child with the disease. Ultimately this means that only about 1 in 100,000 to 1 in 200,000 infants are born with GM1 gangliosidosis each year. However, the exact incidence varies depending on the source of the estimate. Newborn screening for GM1 is not yet universal, but advocacy efforts to add GM1 to newborn screening panels are ongoing. Learn more about newborn screening for GM1.
Subtypes & Symptoms
There are four subtypes of GM1 gangliosidosis: Infantile (Type 1), Late Infantile (Type 2a), Juvenile (Type 2b), and Adult onset (Type 3). Symptoms of GM1 gangliosidosis vary from person to person and type to type. Those with GM1 gangliosidosis may not experience the same symptoms as another patient with the same type of GM1, and the features of the different types can overlap to a great degree. Some symptoms that are typical across all types include: ataxia, dystonia, hypotonia, seizures, and loss of skills.
Symptoms of GM1 Gangliosidosis
Symptoms of GM1 gangliosidosis vary by subtype and by individual, but several features are common across all forms of the disease. Early signs may be subtle and are often mistaken for other conditions, which can delay diagnosis.
Common symptoms across all subtypes include:
- Hypotonia (low muscle tone)
- Ataxia (loss of coordination and balance)
- Dystonia (involuntary muscle contractions)
- Seizures
- Progressive loss of motor and cognitive skills
- Speech and language regression
- Visual impairment
Some children with the infantile form (Type 1) may also show coarsened facial features, enlarged organs (hepatosplenomegaly), and a cherry-red spot on the retina. Symptoms, severity, and rate of progression differ significantly across the four subtypes. Learn more about the subtypes.
History of GM1 Gangliosidosis
Tay-Sachs (GM2 gangliosidosis) was first identified in the late 1800s. It was not until the 1930s that Tay-Sachs was characterized as an “inborn error of metabolism,” and not until the 1960s that the term “ganglioside” was introduced and GM1 gangliosidosis was distinguished as its own disease.
GM1 Gangliosidosis is also known as Landing disease. Dr. Landing et al first identified the disease in 1964, calling it familial neurovisceral lipidosis.
Other Names and Identifiers
- Landing disease
- Norman-Landing disease
- Gangliosidosis-GM1 beta-galactosidase-1 deficiency
- Pseudo-Hurler disease
Life Expectancy for Those Living with GM1
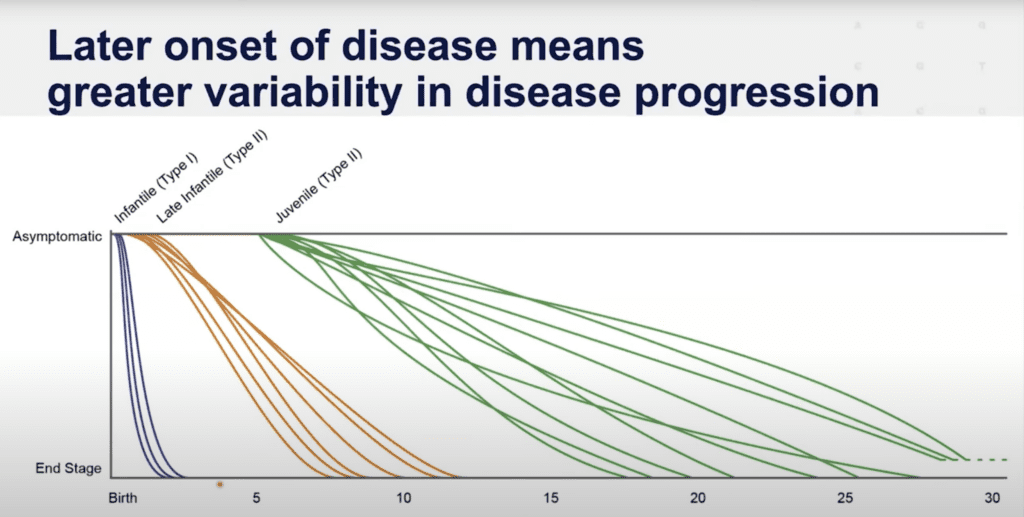
GM1 gangliosidosis is highly variable. Although distinct subtypes exist, the disease is a spectrum based on the amount of residual enzyme activity. Families often ask why life expectancy is so difficult to predict. No two cases are exactly the same, and many variables factor in, including day-to-day medical management.
Families make different choices about intervention levels (such as tracheostomy, feeding tube, or clinical trial participation), which affects outcomes. Published life expectancy data also varies by study, since researchers see different patient populations. For example, the Middle East and Malta are known for particularly severe infantile presentations of GM1, while Japan has a higher documented incidence of Type 3 adult-onset GM1. Life expectancy for adult-onset GM1 requires further study and is not reflected in the diagram below.
Dr. Cynthia Tifft from NIH presented the following diagram in the GM1 Externally-Led Patient-Focused Drug Development meeting with FDA in 2022.
Treatment and Research
As of 2026, there is no approved treatment or cure for GM1 gangliosidosis. Symptom management remains the primary approach and may include anti-seizure medications, muscle relaxants for dystonia, physical therapy, occupational therapy, speech therapy, nutritional support, and palliative care. Research into disease-modifying therapies is actively progressing. The most advanced approaches include AAV gene therapy targeting the GLB1 gene defect, small molecule therapies such as pharmacological chaperones, and enzyme replacement therapy (ERT), which Cure GM1 Foundation is directly funding. The pipeline has expanded significantly in recent years, and there is real reason for optimism. You can help advance this research by donating.

Finding treatments and a cure for GM1 is possible. Researchers and drug developers need the time, support, and resources to get there. You can help advance this research by donating.
If you or a loved one are affected by GM1, you can find support in our Facebook groups. If you or your family are interested in participating in clinical trials and research, please contact us and we can answer questions. You can also see our natural history studies pages.
We invite and encourage anyone interested to take action to further our cause. There are many ways you can use your time and resources to make a difference. Your actions have impact.
To learn more, you can also visit our GM1 Gangliosidosis Facts page.
Frequently Asked Questions
What is GM1 Gangliosidosis?
GM1 Gangliosidosis is a rare inherited genetic disorder caused by a mutation in the GLB1 gene, which results in a deficiency of the enzyme beta-galactosidase. Without this enzyme, a substance called GM1 ganglioside accumulates in cells, primarily neurons, causing progressive neurodegeneration. It is fatal and currently has no approved cure or treatment.
What causes GM1 Gangliosidosis?
GM1 Gangliosidosis is caused by mutations in the GLB1 gene that lead to a deficiency of the enzyme beta-galactosidase. This is a lysosomal storage disorder, meaning the enzyme deficiency causes harmful substances to build up inside cells. The accumulation of GM1 ganglioside in neurons eventually leads to cell death and the progressive deterioration of the nervous system.
How is GM1 Gangliosidosis inherited?
GM1 Gangliosidosis is inherited in an autosomal recessive pattern. This means a child must inherit one mutated copy of the GLB1 gene from each parent to develop the disease. Parents who each carry one copy of the mutated gene are called carriers and typically show no symptoms themselves. When both parents are carriers, each pregnancy carries a 25% chance of an affected child.
How rare is GM1 Gangliosidosis?
GM1 Gangliosidosis is an ultra-rare disease, affecting approximately 1 in 100,000 to 1 in 200,000 infants born each year. Only about 1 in 250 people in the general population carry a single copy of the disease-causing GLB1 mutation.
What are the subtypes of GM1 Gangliosidosis?
There are four subtypes. Type 1 (Infantile) is the most severe, with onset in the first months of life. Type 2a (Late Infantile) has onset between 7 months and 3 years. Type 2b (Juvenile) has onset between 3 and 10 years. Type 3 (Adult onset) is the mildest form and may not become apparent until adulthood. All subtypes are progressive and currently have no approved treatment.
Is there a treatment or cure for GM1 Gangliosidosis?
As of 2026, there is no approved treatment or cure. Symptom management through medications, physical therapy, and palliative care remains the current standard of care. However, research is advancing. Cure GM1 Foundation is directly funding development of an enzyme replacement therapy (ERT), and additional approaches including gene therapy and small molecule treatments are under active investigation.
Ways to Give – Your Support Matters
RaiseRight | Walmart Spark Good | Facebook Fundraisers | Donate | Set up a Recurring Donation
Visit our Take Action page for more ways to support our community